
Organo-Katalyse
Asymmetrische Strecker-Reaktion mit Chinin
Es wurde eine einfache organokatalytische enantioselektive Synthese von N-Arylsulfonyl-α-aminonitrilen von den entsprechenden α-Amidosulfonen entwickelt. Diese Chinin-katalysierte Strecker-Reaktion liefert die cyanierten Produkte in guten Ausbeuten und Enantioselektivitäten.
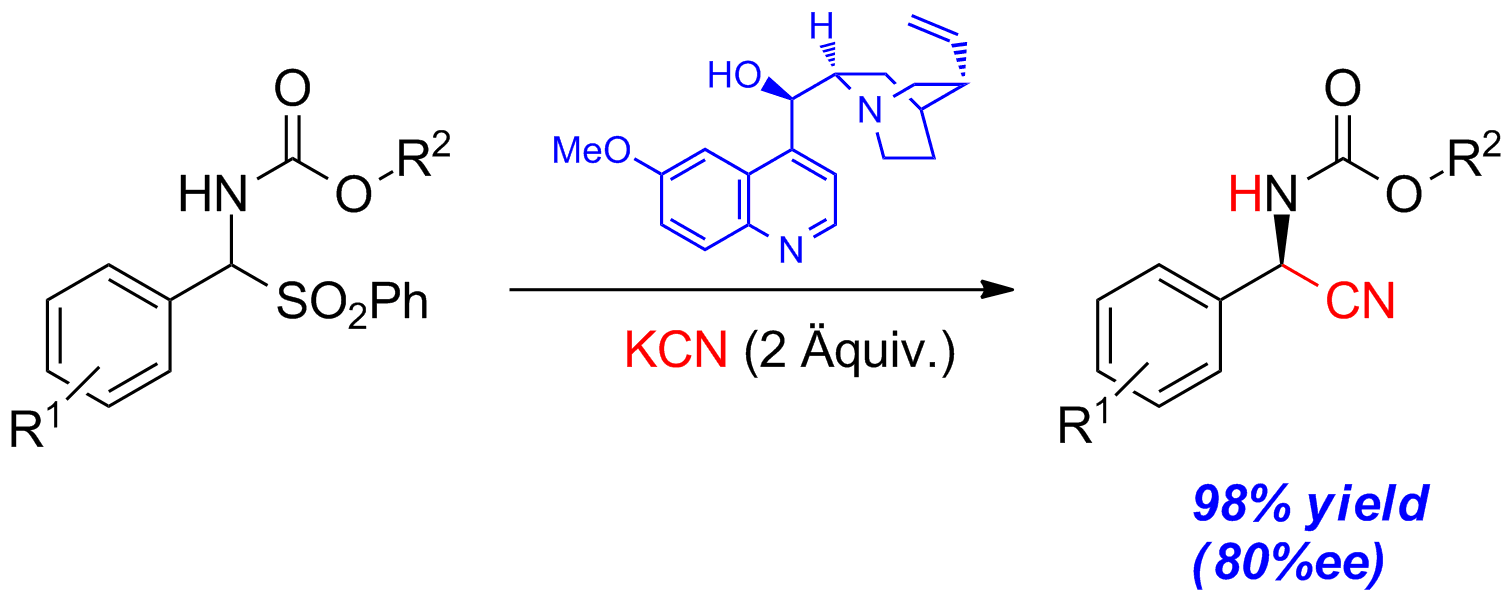
Schema 1: Organokatalytische Strecker-Reaktion mit Chinin.
Um hochtoxisches und leicht flüchtiges HCN zu vermeiden, wurde Kaliumcyanid verwendet, welches auf Grund seiner leichteren Handhabung ein weiter verbreitetes Cyanierungsmittel ist. In dieser Variante besitzt KCN zwei Funktionen. Zum einen fungiert es als Base und generiert dadurch in situ das freie N-Carbamoylimin durch Deprotierung des Ausgangsmaterials und anschließender Eliminierung von Sulfinat. Zum anderen setzt es intermediär HCN frei, welches anschließend als Cyanierungsmittel fungiert. In Kombination mit einem geeigneten chiralen Katalysator können so die Strecker-Produkte enantioselektiv erhalten werden.
Asymmetrische Aminierung von Aldehyden
Ein effizienter Zugang zu konfigurationsstabilen α,α-disubstituierten α-Aminosäuren ist sehr wünschenswert. Von simplen und einfach zugänglichen racemischen Aldehyden können α-sulfamidierte Aldehyde in guten Ausbeuten und Enantioselektivitäten erhalten werden (Schema 2).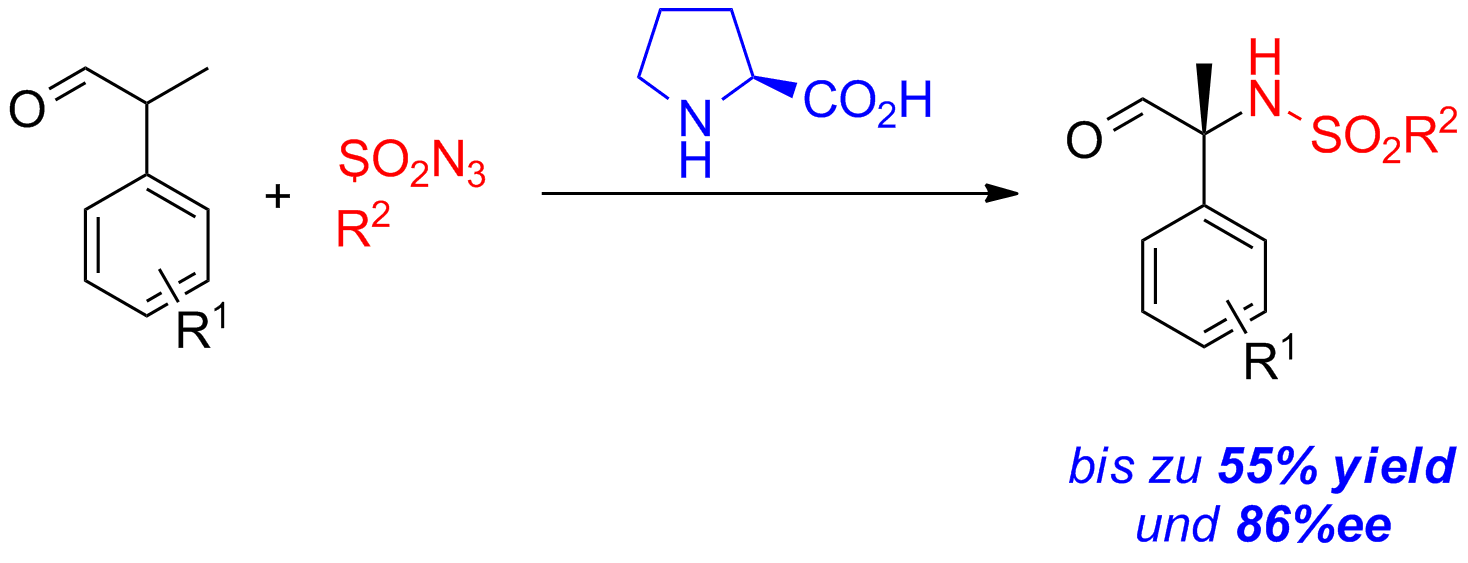
Schema 2: Asymmetrische α-Sulfamidierung von α-verzweigten Aldehyden.
Die erhaltenen Produkte können mit zwei weiteren Schritten in die entsprechenden unnatürlichen Aminosäuren überführt werden.
Unter Anwendung von Mikrowellen können gute bis zu exzellente Ausbeuten unter signifikant reduzierten Reaktionszeiten erhalten werden. Die N-Funktionalität wird in diesem Fall durch ein Diazoderivat (DEAD) eingeführt. Es können auf diese Weise α-aminierte Produkte mit bis zu 86%ee erhalten werden (Schema 3).
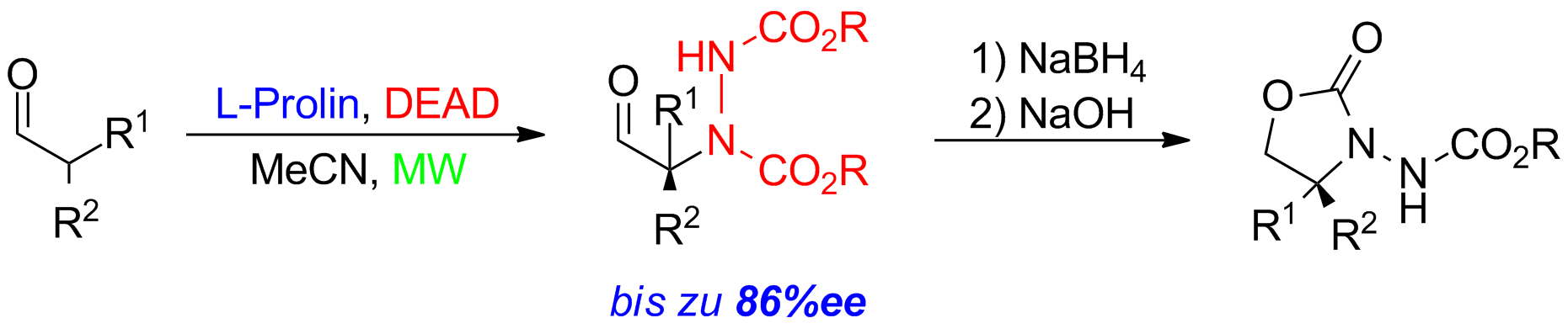
Schema 3: Prolin-katalysierte asymmetrische Aminierung von α-verzweigten Aldehyden.
Die erhaltenen Produkte können weiter zu den synthetisch wertvollen Oxazolidinone überführt werden. Aber auch die Überführung in α-Aminoalkohole, Carbonsäuren und α-alkylierten Phenylglycin-Derivaten ist möglich.
Asymmetrische Diels-Alder-Reaktion
Ein Schlüsselschritt in der Totalsynthese von Δ9-Tetrahydrocannabinol stellt die organokatalytische Diels-Alder-Reaktion dar. In Vorarbeiten wurden verschiedene MacMillan-Katalysatoren getestet, wobei bereits hervorragende Enantioselektivitäten (bis zu 98%ee) erreicht werden konnten (Schema 4). 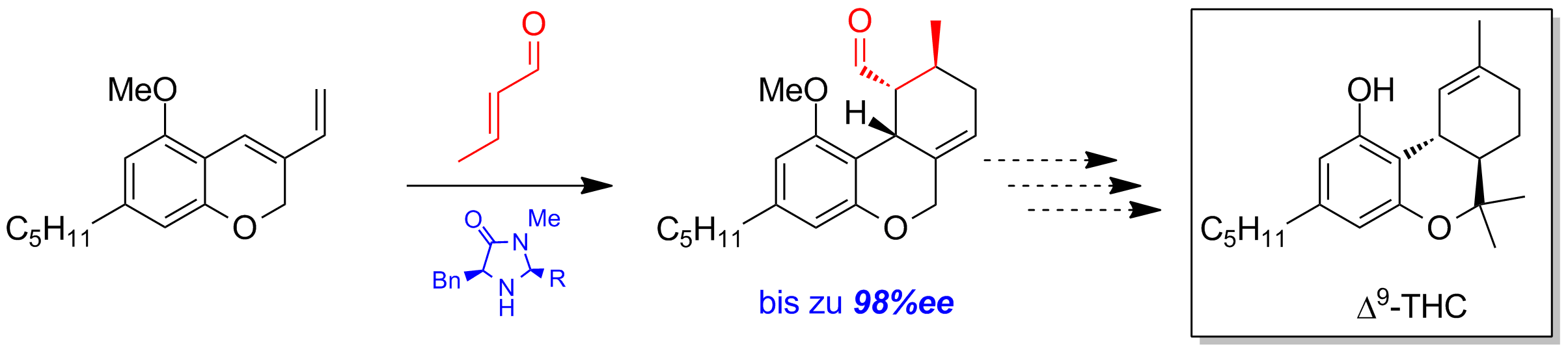
Schema 4: Asymmetrische Diels-Alder mit MacMillan-Katalysator.
Domino-Oxa-Michael-Aldol-Reaktion / Formale Totalsynthese von 4-Dehydroxydiversonol
Die basenkatalysierte Dominoreaktion von Salicylaldehyd 1 mit α,β-ungesättigten Aldehyden 2 wird seit kurzem in Hinblick auf die Totalsynthese von Naturstoffen näher untersucht. In Abhängigkeit der Reaktionsbedingungen werden Chromene 3 durch eine Oxa-Michael-Aldol Reaktion oder Lactol 4 durch eine vinyloge Aldol-oxa-Michael Reaktion erhalten (Schema 5).
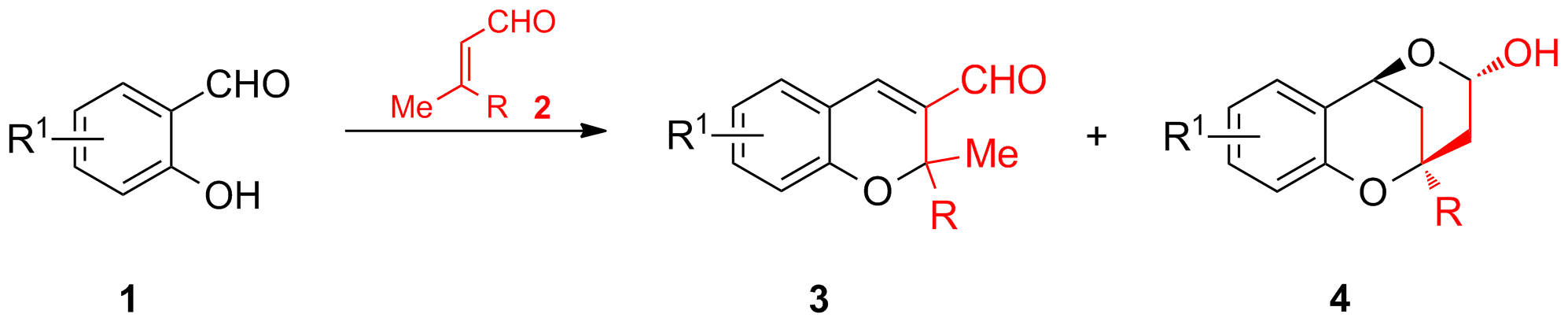
Schema 5: Mögliche Produkte bei der Domino-Oxa-Michael-Aldol-Reaktion.
Mit dieser Methode ist es möglich, Salicylaldehyd 1a (erhältlich aus Orcinol über 3 Stufen) in das Lactol 4a in hohen Enantiomerenreinheiten unter Verwendung des Jørgensen-Katalysators5 zu überführen. Über mehrere Schritte kann 4a in das Xanthenonderivat 6 überführt werden, welches anschließend nach Tietze et al. zu 4-Dehydroxydiversonol (7) liefert (Schema 6).
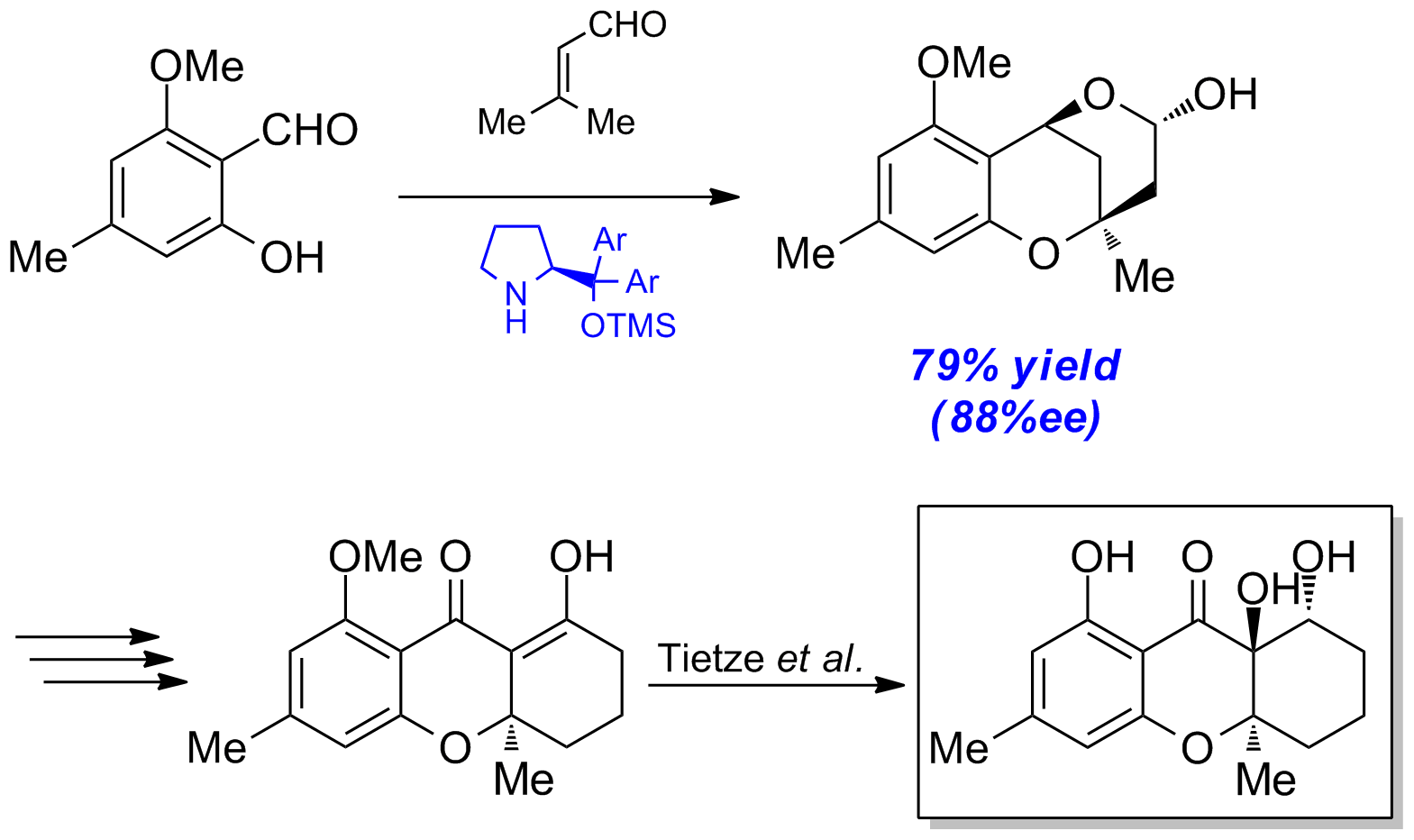
Schema 6: Formale Totalsynthese von 4-Dehydroxydiversonol.
Metall-Katalyse
Asymmetrische Synthese mit einem Rh-PCP-Katalysator
Die Übergangsmetall-katalysierte Zersetzung von Diazoverbindungen bietet ein einfaches, skalierbares und vielseitiges Werkzeug für den Zugang zu Cyclopropanen.[11] Obwohl Rhodiumcarboxylat-Komplexe die attraktivsten Katalysatoren in Bezug auf Effizienz und Zugänglichkeit für den Aufbau von Cyclopropan- und Cyclopropenringen sind, bleibt eine wesentliche Einschränkung ihre mangelnde Selektivität gegenüber der b-Hydrid-Migration.[12]
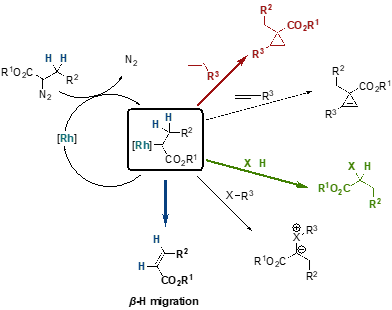
Schema 7: Mögliche Reaktionen und Mechanismus.
[2.2]Paracyclophancarbonsäure und Derivate davon werden synthetisiert. Der entsprechende Rhodium-Paddlewheel-Komplex wird in der Cyclopropanierungsreaktion von terminalen, a,a - und a,b -disubstituierten Alkenen mit a-Alkyl-a-diazoestern eingesetzt. Die einzigartige Massivität von [2.2]Paracyclophan ermöglicht eine chemoselektive Cyclopropanierung mit hoher Ausbeute bei Raumtemperatur, während seine inhärente planare Chiralität derzeit in enantioselektiven Cyclopropanierungsreaktionen untersucht wird.[13]
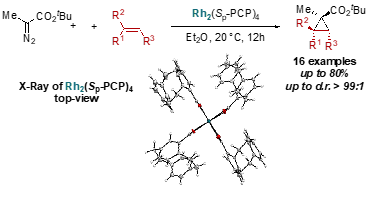
Schema 8: Getestete Reaktionen.
Asymmetrische Michael-Addition
Unter Cometall-freien Bedingungen können α,β-ungesättigte Aldehyde als Substrate in einer asymmetrischen Michael-Addition eingesetzt werden. Hierzu werden lediglich 2 mol% des auf [2.2]Paracyclophan basierenden N,O-Liganden benötigt. Auf diese Weise können die gewünschten Michael-Addukte selektiv in exzellenten Enantiomerenüberschüssen erhalten werden (Schema 7).
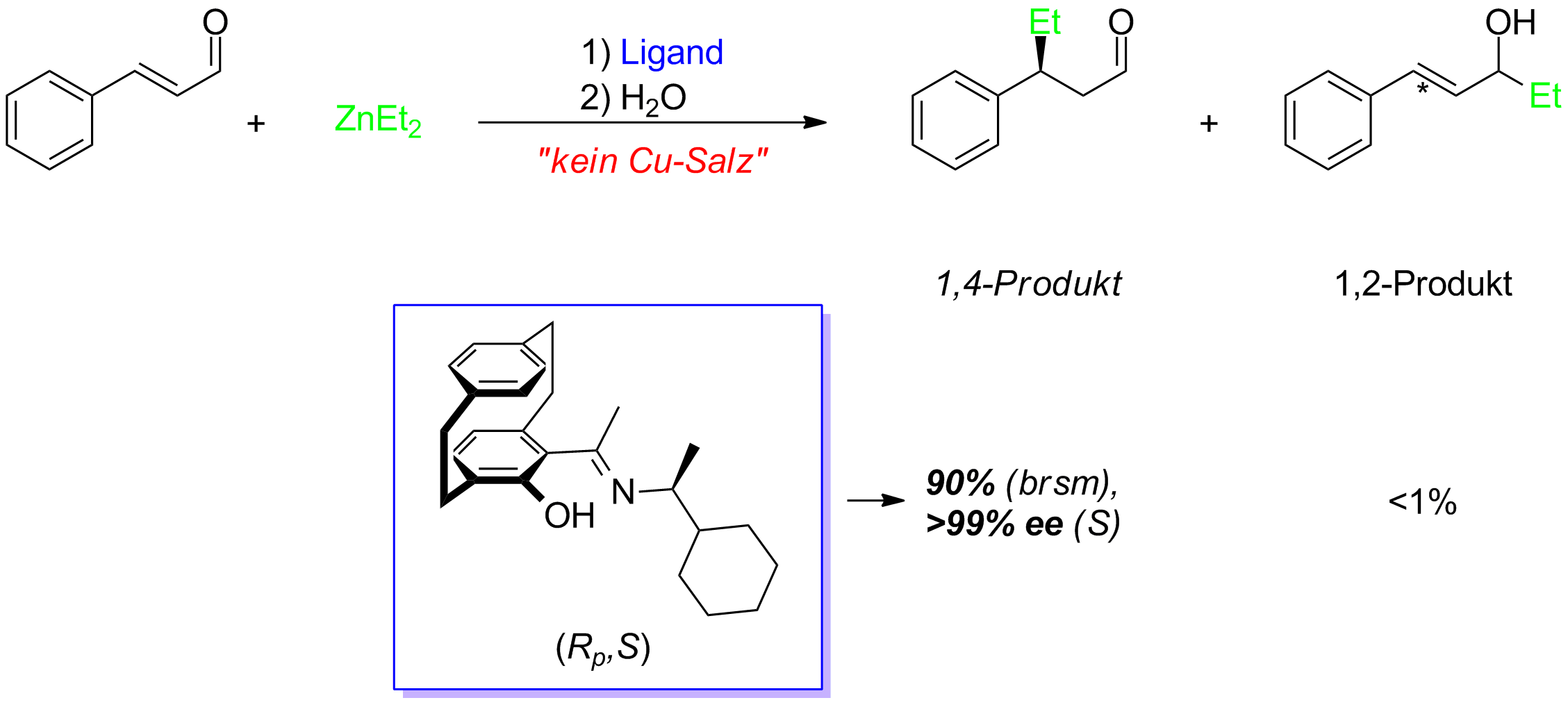
Schema 9: Asymmetrische Michael-Addition unter Cometall-freien Reaktionsbedingungen.
Referenzen
[1] T. Baumann, H. Vogt, S. Bräse, Eur. J. Org. Chem. 2007, 266–282. The proline-catalyzed asymmetric amination of branched aldehydes.
[2] J. Toräng, S. Vanderheiden, M. Nieger, S. Bräse, Eur. J. Org. Chem. 2007, 943-952. Synthesis of 3-Alkylcoumarins from Salicylaldehydes and α,β-unsaturated Aldehydes utilizing Nucleophilic Carbenes: A New Umpoled Domino Reaction
[3] R. Reingruber, S. Bräse, Chem. Commun. 2008, 105-107. 1,2-Addition of Trialkylaluminium Reagents on N-Diphenylphosphinoyl-ketimines in the Absence of Any Additional Reagents
[4] T. Baumann, M. Bächle, C. Hartmann, S. Bräse, Eur. J. Org. Chem. 2008, 2207-2212. Thermal Effects in the organocatalytic asymmetric α-amination of disubstituted aldehydes with azodicarboxylates: a high-temperature organo-catalysis (Cover picture)
[5] R. Reingruber, S. Vanderheiden, A. Wagner, M. Nieger, T. Muller, M. Es-Sayed, S. Bräse, Eur. J. Org. Chem. 2008, 3314-3327. 1-Aryl-3,3-diisopropyltriazenes: An Easily Accessible and Particularly Stable Class of Triazenes Towards Strong Basic and Lewis Acid Conditions
[6] S. Ay, M. Nieger, S. Bräse, Chem. Eur. J. 2008, 14, 11539-11556. Co-Metal-Free Enantioselective Conjugate Addition Reactions of Zinc Reagents
[7] N. Volz, M. C. Bröhmer, S. Bräse, Synlett 2009, 550-553. An Organocatalytic Sequence towards 4a-Methyl Tetrahydroxanthones: Formal Synthesis of 4-Dehydroxydiversonol
[8] R. Reingruber, S. Vanderheiden, T. Muller, M. Nieger, S. Bräse, Tetrahedron Lett. 2009, in press (invited for spezial issue: 50 years of Tetrahedron publications). Efficient synthesis of 3-acylbenzo[1,2,3]triazenes
[9] R. Reingruber, T. Baumann, S. Dahmen, S. Bräse, Adv. Synth. Catal. 2009, in press. Use of the Chiral Pool – Practical Asymmetric Organocatalytic Strecker Reaction with Quinine
[10] M. C. Bröhmer, N. Volz, S. Bräse, Synlett 2009, in press. Rhodium-catalyzed decarbonylation of functionalized 3-formylchromenes: An (asymmetric) sequence for functionalized chromenes like deoxycordiachromene.
[11] C. Ebner, E. M. Carreira, Chem. Rev. 2017, 117, 11651-11679.
[12] T. L. Sunderland, J. F. Berry, Dalton Trans. 2016, 45, 50-55.
[13] C. Zippel, Z. Hassan, M. Nieger, S. Bräse, Adv. Synth. Catal. 2020.