
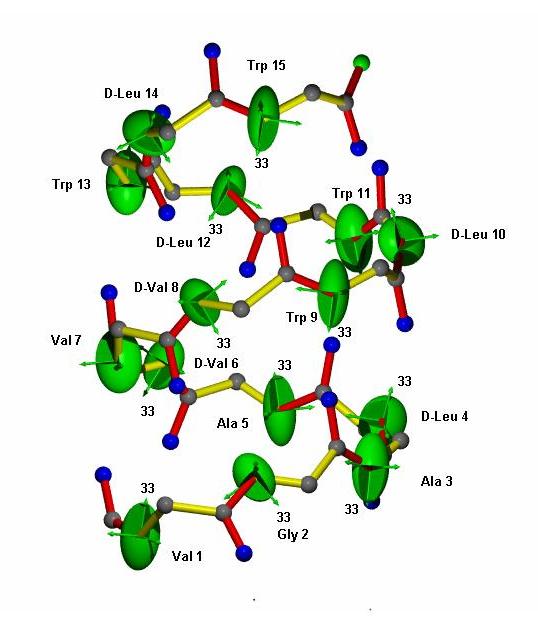
Computational Chemistry
The measured experimental anisotropic parameters contain valuable structural information, which can be used to access various aspects of the assignment of the constitution, configuration, conformation and enantiomeric excess of the organic molecules. A more or less trivial case for the use of the anisotropic parameters is the class of rigid molecules. However, the majority of organic molecules with potential applicability in biological systems contain different degree of flexibility, which fails to be analysed with the static models.
Time averaged molecular dynamics simulations with orientational constraints (MDOC), as implemented in the program COSMOS, can be applied for both classes of organic molecules. The current implementation of the program has been tested for the use of residual dipolar couplings (RDCs). Potentially it allows the use of scalar couplings and NOE distances as constraints during the molecular dynamics run.
Bond Polarization Theory (BPT) - an Extreme Fast Method for the Calculation of Molecular Properties
A new quantum chemical method for the calculation of molecular properties, the bond polarization theory (BPT), was developed within the last years. The BPT was the starting point for the development of a new semi-empirical technique for the calculation of atomic charges and NMR chemical shifts. The BPT method provides a very fast access to chemical shifts, and hence is well suited for the application in molecular mechanics and dynamics simulations. Using the BPT calculation of 13C and 15N chemical shifts and their gradients with respect to coordinates opens the possibility to refine protein structures with CS target functions. A great advantage of the method is that calculations even for very large molecular systems can be performed within errors bounds similar to post Hartree-Fock ab initio calculations.
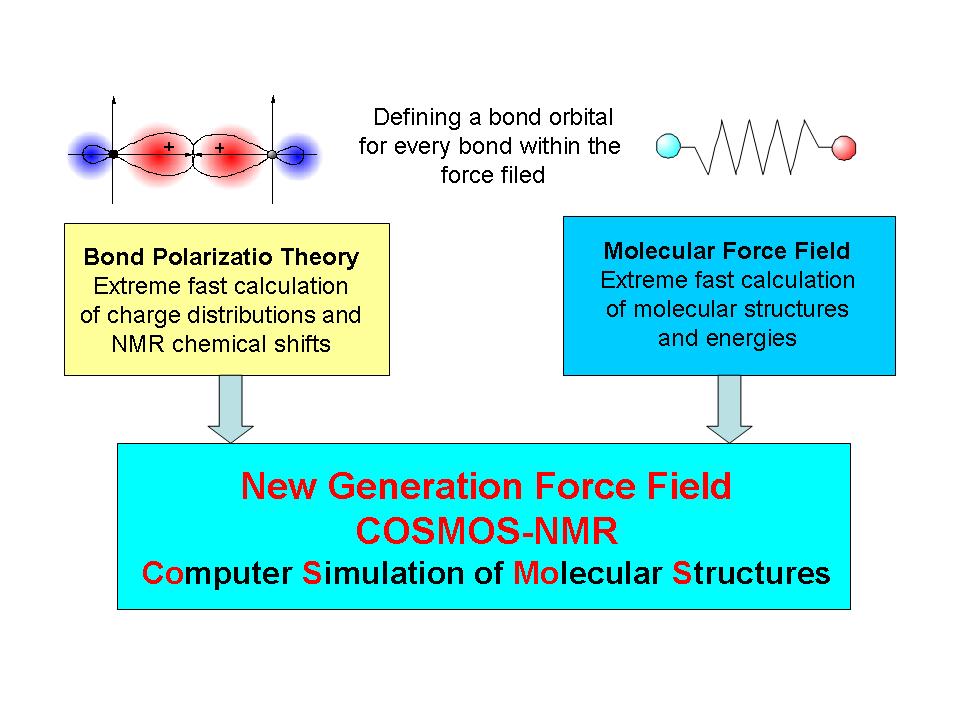
Development of the QM/MM Force Field COSMOS-NMR
A new force field was designed to implement a calculation of Coulomb interactions with fluctuating atomic charges. Using BPT derived atomic charges it is possible to include all mutual polarizations into the term for the electrostatic interaction. The primary goal of this new force field is the better description of the intermolecular interactions of molecular systems. Isotropic and anisotropic NMR parameters can be introduced into force field calculation to simulate NMR experiments and refine structures from NMR data.
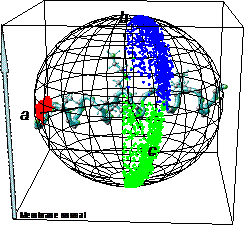
Molecular Dynamics with Orientational Constraints (MDOC)
Molecules in oriented samples like bio-membranes and polymers show a directional dependence of NMR parameters like dipolar couplings or chemical shifts. The most prominent property that can be observed is the residual dipolar coupling (RDC) and structures can be elucidated using RDCs. We developed a new molecular dynamics method (MDOC) were orientational constraints are driving molecular rotations and reorientations.