
Anisotropic Parameters in Liquid State NMR
NMR structure analysis is the main technique used for the determination of configuration, conformation, and constitution of organic compounds. Classical procedures are based on the combined use of chemical shifts, spin-spin coupling constants, and nuclear Overhauser effects (NOEs).
In many cases these data are not sufficient and an alternative approach is required. This is based on the measurement of additional anisotropic NMR parameters in appropriate alignment media. Under such conditions the following anisotropic parameters are accessible: residual chemical shift anisotropy (RCSA), residual dipolar couplings (RDCs) and residual quadrupolar couplings (RQCs).
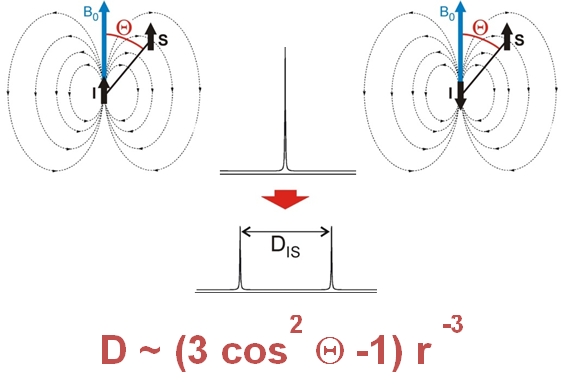
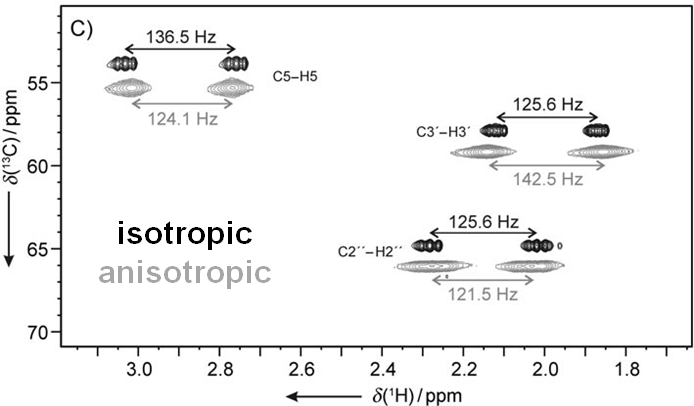
Residual dipolar couplings depend on the internuclear distance between the two NMR active nuclei (r) and the angle of the internuclear vector with respect to the outer magnetic field. Knowledge of RDCs allows the determination of the molecule’s alignment relative to an external reference, thus correlating also distant parts of the molecule. The RDCs have been used to study the conformation, configuration, constitution, and enantiomeric distinction of small-to-medium-sized organic molecules.
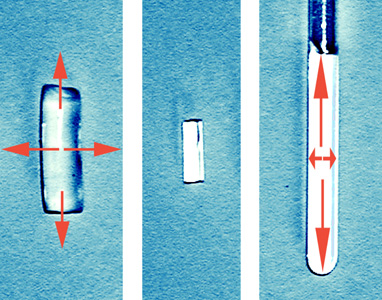
The successful measurement of RDCs requires suitable alignment media which allow proper scaling and solvents range. For organic molecules usually two types of media are used: liquid crystalline phases and stretched polymer gels. A number of polymer gel based alignment media for various solvents has been developed in our group and currently we are working on further optimisation of the alignment media.

The stretching apparatus is an easy way to scale the alignment strength and as well measure RDCs at various alignments with one sample.
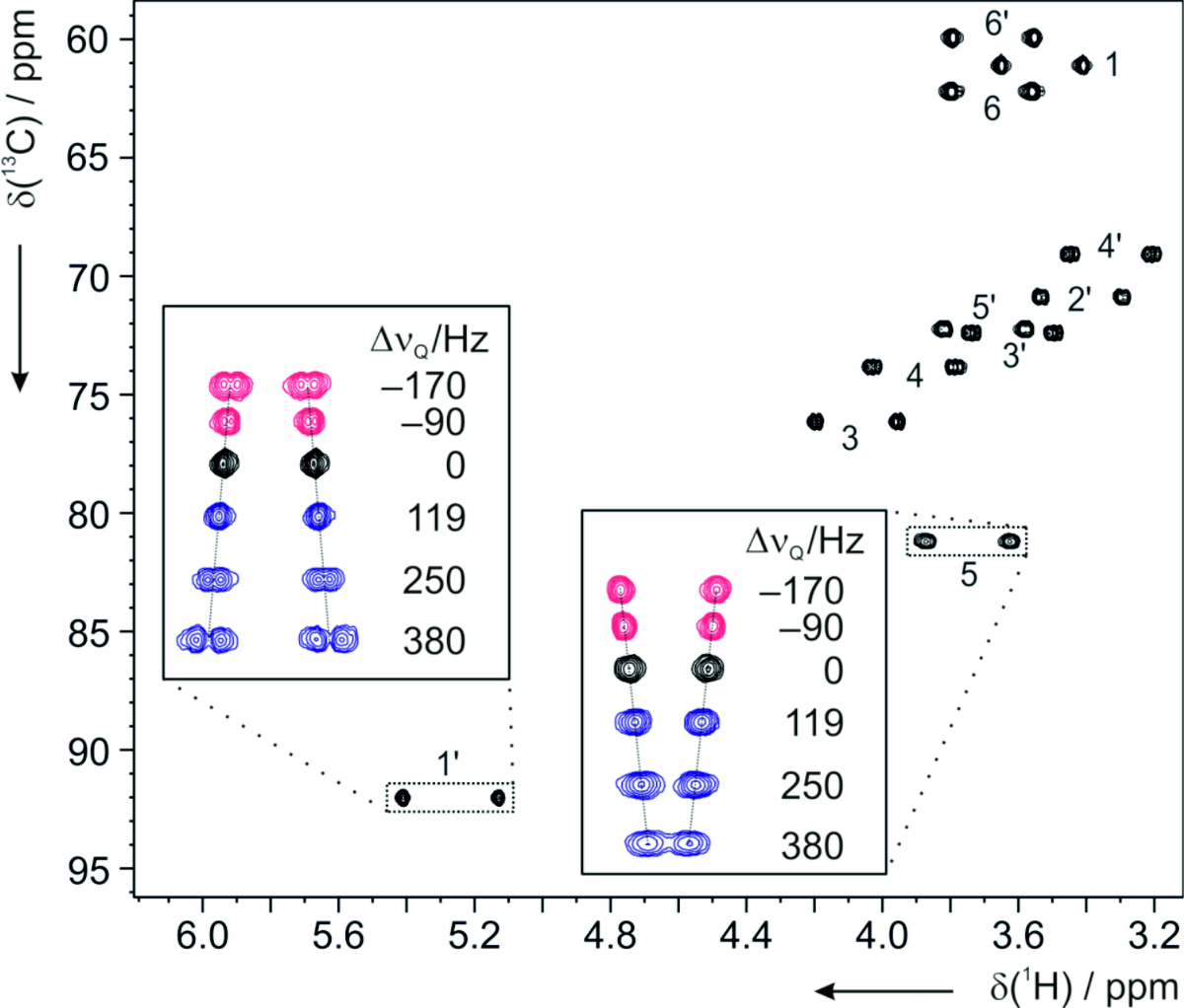
The experimental RDCs are further used to solve different structural problems. During RDC analysis, an alignment tensor is calculated for a sterically fixed orientational model. Agreement between experimental and back calculated RDCs allows to solve the constitution, to assign the configuration, and the conformation of the molecule. The majority of the organic molecules we have recently been working with, contains some certain degree of flexibility. In such cases, the straightforward approach, which assumes a single rigid conformation, fails. Therefore, we have started to evaluate our experimental RDCs with a specialized molecular dynamics approach (see more in the Computational chemistry).